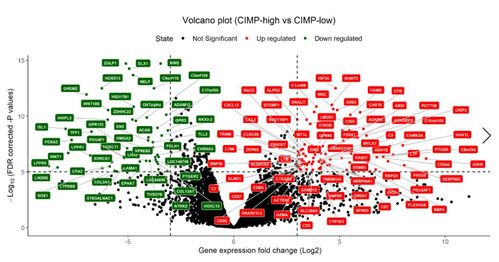
作为一名热爱生物信息学的科研小白,今天我终于鼓起勇气深入研究了转录组分析中的核心内容——GO及KEGG富集分析及其可视化。这不仅是一次技术上的突破,更是一场思维上的飞跃!
在转录组数据分析中,差异基因的筛选只是第一步,更重要的是对这些差异基因的功能进行解读。而GO(Gene Ontology)和KEGG(Kyoto Encyclopedia of Genes and Genomes)富集分析正是这一过程的核心工具。
什么是GO和KEGG富集分析?
简单来说,GO富集分析可以帮助我们了解差异基因主要参与哪些生物学功能、细胞组分以及分子功能。而KEGG则侧重于揭示这些基因在代谢通路或信号通路中的作用。两者结合使用,可以全面解析基因表达变化背后的生物学意义。
从理论到实践:如何进行GO与KEGG富集分析
首先,我们需要准备一份差异基因列表,这是整个分析的基础。接着,通过R语言中的clusterProfiler包,我们可以轻松完成GO和KEGG富集分析。以下是我的具体操作步骤:
- 加载必要的R包:包括clusterProfiler、org.Hs.eg.db等。
- 导入差异基因数据:确保数据格式正确,通常包含基因ID和log2FoldChange值。
- 运行GO富集分析:使用enrichGO函数,设置参数如ontology(BP、CC、MF)和pvalueCutoff。
- 运行KEGG富集分析:利用enrichKEGG函数,指定物种代码。
可视化:让数据说话
完成富集分析后,接下来就是展示成果的时间了!条形图和气泡图是两种最常用的可视化方式。条形图可以清晰地展示每个GO term或KEGG pathway的富集程度,而气泡图则更加直观地反映了富集结果的多重属性。
以下是我在R中绘制条形图和气泡图的代码片段:
# 条形图绘制
barplot(gene_GO, showCategory=10)
# 气泡图绘制
dotplot(gene_KEGG, showCategory=10)通过这些图表,我能够一目了然地看到哪些通路或功能被显著富集,从而为后续实验设计提供了明确的方向。
总结与感悟
经过这次实践,我对转录组分析有了更深的理解。GO和KEGG富集分析不仅是数据挖掘的利器,更是连接基因表达与生物学意义的桥梁。未来,我将继续探索更多高级的分析方法,比如GSEA(Gene Set Enrichment Analysis),以进一步提升自己的科研能力。
如果你也对转录组分析感兴趣,不妨从GO和KEGG富集分析开始吧!相信你也会像我一样,在这个过程中收获满满的知识与成就感。


发表评论 取消回复